Innerhalb der Erbinformation (DNA) bilden 4 Basen in vielfaltigen Kombinationen den genetischen Code des Menschen: Adenin, Guanin, Cytosin und Thymin.
Gene codieren zu gleichnamigen Proteinen (Eiweißen). Das kann man sich vorstellen als ob eine Kopiermaschine immer wieder über das Gen fährt, den Code abliest und mit diesen Informationen das Protein baut. Wie Handwerker den Bauplan lesen wenn sie ein Haus bauen. Trägt das Gen MECP2 Fehler (trägt also eine Mutation), übernimmt auch das Protein (Eiweiß) MECP2 diese Fehler aus dem mutierten Code des Gens MECP2. Ist der Bauplan falsch, wird das Haus schief.
Das Protein MECP2
hat im Körper eine Funktion ähnlich einem zentralen Schalter.
Da es sehr weit oben sitzt innerhalb der Schaltmechanismen regelt es die An- und
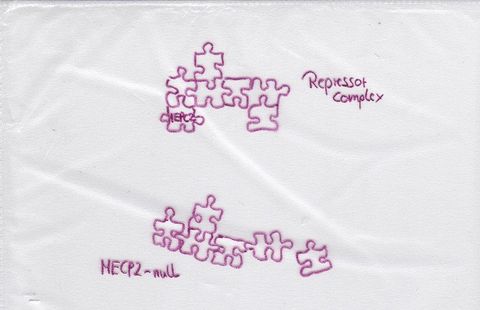
Abschaltvorgänge anderer Proteine. Es wird als multifunktionales Gen gesehen.Optisch können wir uns ein Puzzle vorstellen, das durcheinander gerät, wenn MECP2 nicht korrekt funktioniert.
MECP2 ist ein Stillleger und hauptsächlich in Nervenzellen aktiv. Ist das MECP2 gestört ist auch die neuronale Verknüpfung gestört. Optisch können wir uns die nicht ausreichend verknüpften Nervenzellen als struppige Weihnachtsbäume vorstellen. Sie weisen geringere und viel weniger verästelte Zweige auf als buschige üppige Weihnachtsbäume.
Interessant in diesem Zusammenhang ist, dass auch Stress und Angst ähnliche Effekte erzeugen. Ebenso entsteht ein schlechtes neuronales Verknüpfungsmuster.
Eine herabgesetzte Schmerzempfindlichkeit ist ein Anzeichen für mangelnde Verknüpfung der Neuronen. Die Ausprägung und das Auftreten der gestörten Verknüpfung sind beim Rett-Syndrom unterschiedlich.
Verlauf oft unterschiedlich
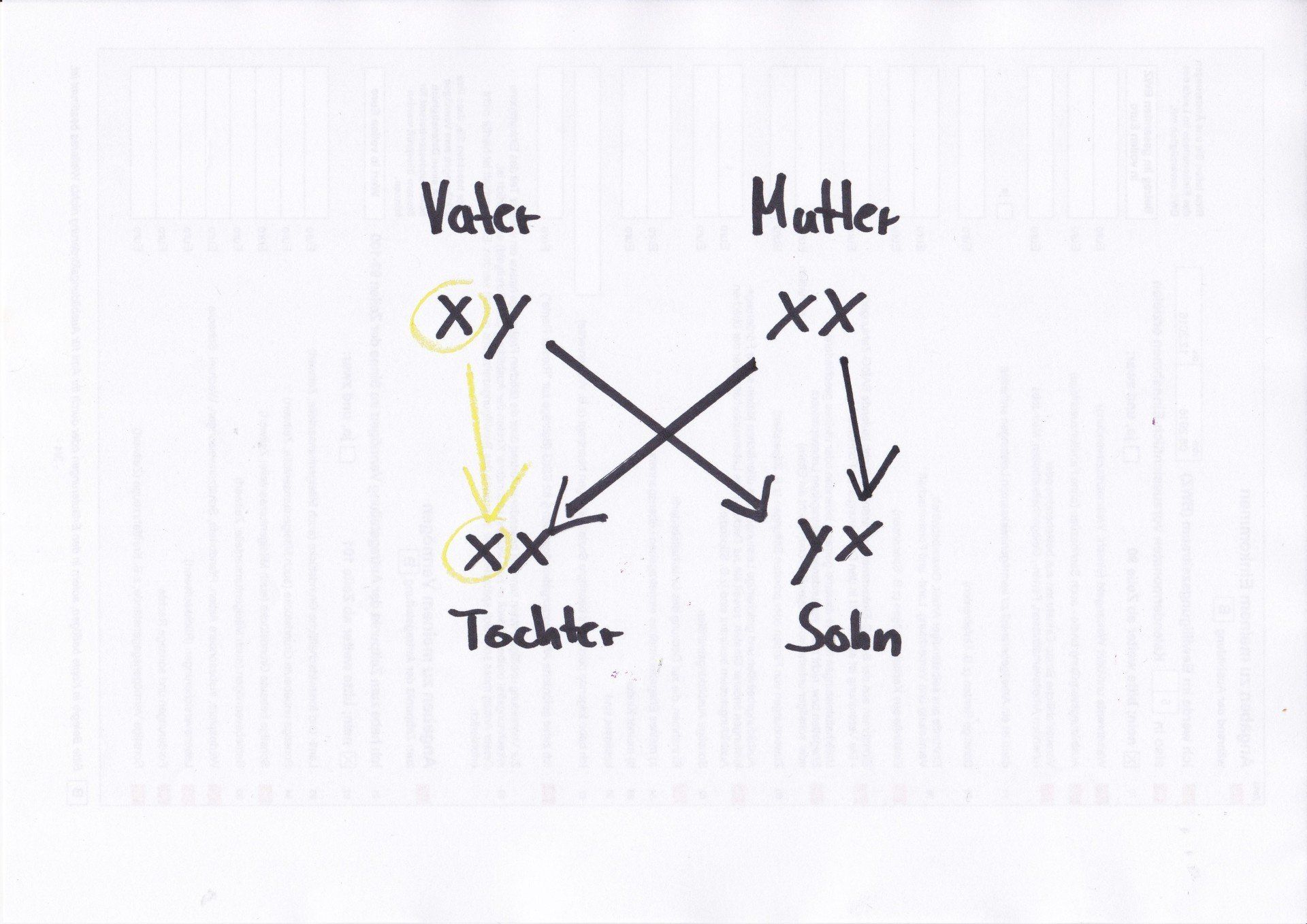
Die Vorgänge, die zur Ausbildung der Krankheitssymptome führen, sind äußerst komplex. Da jedes Mädchen über zwei X-Chromosomen verfügt, eines vom Vater, eines von der Mutter, aber pro Zelle nur ein X-Chromosom benötigt wird, kommt es in einer willkürlichen Ausprägung zum Abschalten eines der beiden X-Chromosomen. Statistisch werden dabei im Verhältnis 50:50 entweder das kranke oder das gesunde Chromosom abgeschaltet bzw. eingeschaltet. Praktisch kann das Verhältnis aber deutlich varieren. Vielleicht ist es 70:30, 40:60 etc. krank zu gesund. Das könnte den oft sehr unterschiedlichen Verlauf beim Rett-Syndrom
erklären. Dieses Verhältnis bleibt so, da alle Tochterzellen einer Zelle dieselbe X-Inaktivierung tragen.
Bei Jungen, die nur ein X-Chromosom besitzen, ist in jeder Körperzelle das Rett-Gen aktiviert. Das könnte den besonders schweren Verlauf des Rett-Syndrom bei Jungen erklären.
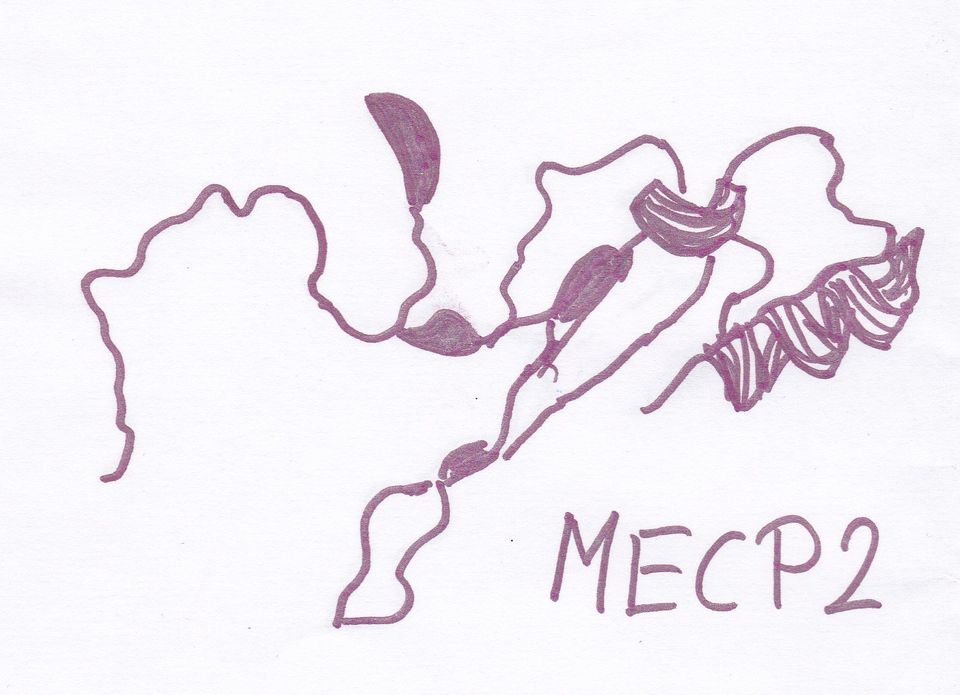
MECP2, 1999 als Ort der Mutation für Rett entdeckt, stellt sich 19 Jahre später äußerst vielgestaltig dar.
Man spricht mittlerweile von einer MECP2 Spektrum-Störung.
über das Rett-Syndrom hinausgehend.
Auch wird das MECP2 Duplikationssyndrom
beschrieben.
Ebenso werden die Symptome von Rett durch mindestens zwei weitere Mutationen in anderen Genen ausgelöst:
weitere Infos zum Thema 2019 Vortrag Rett-Forschung mit Dr. Charlotte Stein, Biologin, Elternhilfe Bayern